Sarah B. Scruggs, Jei Wang, Peipei Ping, UCLA David Geffen School of Medicine, Departments of Physiology, Medicine/Cardiology, and Computer Science, NIH BD2K Center of Excellence for Biomedical Computing at UCLA (HeartBD2K)
 The Cardiovascular BD-HPP Initiative welcomes a new era of precision medicine that capitalizes on future-forward proteome technologies to identify cardiovascular disease mechanisms and therapeutic targets. Proteome signatures have gained recognition as the main driving force in cellular phenotypes, and several extraordinary and quantitative innovations can now elucidate these dynamic fingerprints at unprecedented speed, sensitivity, specificity and scale. Significant advancements that are forging new heights in proteomic analysis include new multiplexed mass spec sample preparation workflows and developments in novel, quantitative phospo- and splice variant proteoform resolution using top-down mass spec technologies.
The Cardiovascular BD-HPP Initiative welcomes a new era of precision medicine that capitalizes on future-forward proteome technologies to identify cardiovascular disease mechanisms and therapeutic targets. Proteome signatures have gained recognition as the main driving force in cellular phenotypes, and several extraordinary and quantitative innovations can now elucidate these dynamic fingerprints at unprecedented speed, sensitivity, specificity and scale. Significant advancements that are forging new heights in proteomic analysis include new multiplexed mass spec sample preparation workflows and developments in novel, quantitative phospo- and splice variant proteoform resolution using top-down mass spec technologies.
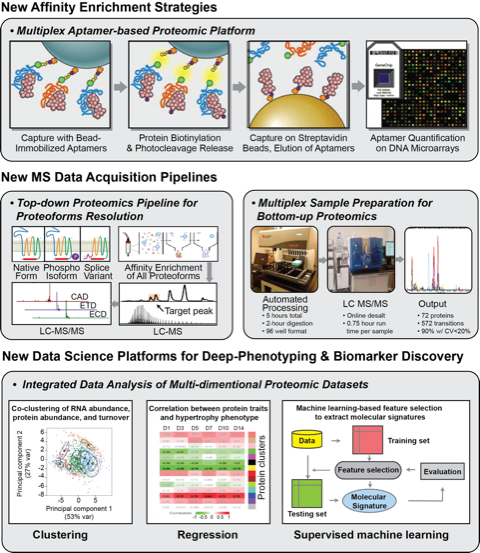
New and exciting data from Ying Ge’s laboratory unveiled the complex proteoform profile of cardiac Troponin T (cTnT), a critical thin filament regulatory molecule of cardiac contraction/relaxation and clinical biomarker of myocardial infarction. Using online liquid chromatography and immunoaffinity purification coupled to high resolution top-down MS and several complementary fragmentation approaches (CAD, ECD, ETD), they were able to unequivocally identify seven unique cTnT splice variants, each in an unphosporylated or mono-phosphorylated form. This study represents the unprecedented power of top-down MS in isolating various intracellular proteoforms that likely elicit distinctive biological functions.
Exciting work by Jennifer van Eyk’s group includes the development of a streamlined, multiplexed workflow that enables the processing of 96 complex biological samples for MS in a matter of 5 hours. This methodology demonstrated remarkable reproducibility for both plasma and serum samples, showcasing CV’s less than 20% and holding up across replicates, days, instruments, and laboratory sites. Overall, this valuable approach will empower the proteomics community with tools to minimize artefactual introductions and enhance the native biological presentation of peptides during MS analysis. Moreover, we are seeing unparalleled developments in multiplexed, aptamer-based technologies which now target nearly 5000 proteins in biofluids. A collaborative study between Robert Gerszten and Lori Jenning’s groups demonstrated the application of this method to identify circulating biomarkers for myocardial infarction in derivation and validation patient cohorts, with orthogonal technical validation using targeted mass spectrometry
Computational innovations incorporating global proteome dynamics into data science platforms have emerged. The integration of transcript abundance, protein abundance, and protein turnover data and the extraction of multi-dimensional molecular signatures affords a 75% gain in discovering disease gene candidates. These recent developments in data science tools will facilitate protein marker discoveries, including clustering as well as signature extraction.
In summary, new technologies are paving the way to resolving comprehensive, spatio-temporal, dynamic maps of cardiovascular and other organelle proteomes that expand and articulate our understanding of how protein networks propel myriad phenotypes. Capitalizing on this knowledge to ultimately reduce the number of cardiovascular disease-related deaths is truly on the horizon.


.png)












