Geoffrey G. Hesketh1 & Anne-Claude Gingras1,2
1Lunenfeld-Tanenbaum Research Institute, Sinai Health System
2Department of Molecular Genetics, University of Toronto
Toronto, Ontario, Canada
Cells can be viewed as functioning in four dimensions: three-dimensional space (structural organization) and one-dimensional time (dynamics). To understand cell function, we must therefore understand how their constituent macromolecules, including proteins, lipids, carbohydrates and nucleic acids, exist in space as well as in time. Here, we discuss the use of proteomics methods to probe such questions.
The era of molecular cell biology (i.e., studying the mechanisms by which molecules orchestrate cell function) was ushered in during the 1950s with the development of two key techniques – cellular electron microscopy and cell fractionation by differential centrifugation. By combining these powerful approaches with existing classical biochemistry methods, critical insight into how cells are organized into distinct membrane-bound organelles (e.g., ER, Golgi, mitochondria, lysosomes) and molecular structures and machines (e.g., chromatin, ribosomes) was gained. Importantly, this led to the realization that these different compartments carry out distinct cellular functions, largely due to the differential partitioning of specific macromolecules to distinct compartments.
In the late 1990s and early 2000s, genome sequencing and accelerated development in mass spectrometry technologies for peptide sequencing set the stage for the new field of proteomics, which was ideally suited to addressing molecular cell biology questions. Rather than analyzing differential cell fractions by low throughput biochemistry methods, the ability to assign proteins en masse to distinct fractions (and therefore distinct cell compartments) became possible. The speed, sensitivity, and resolution of mass spectrometers dramatically improved over the years, and when combined with modern proteomics technologies (including multiplexed quantitation by isotope tagging strategies and improvements in data analysis), spatial proteomics was able to develop into a mature field1.
An inherent limitation to spatial proteomics methods that rely on cell lysis and differential fractionation is that spatial information is captured after both lysis and fractionation have occurred. Lysis necessarily involves some form of membrane disruption (often by mechanical means and the use of detergents) and the fractionation (the stage at which ‘spatial’ information is captured) is carried out under non-native conditions. While the integrity of certain cellular compartments may be maintained during such procedures, this is not universally true for all compartments. Furthermore, it is becoming increasingly appreciated that many cell functions are driven by extensive contacts between distinct organelles2, and these associations are poorly captured by lysis and fractionation approaches.
Methods that bypass the requirement for isolating intact structures prior to identification by mass spectrometry can overcome many of these limitations. The first practical application of proximity-dependent biotinylation followed by mass spectrometry (BioID) was described in 20123; reviewed in4). BioID employs a bacterial biotin ligase (BirA) in which a single point mutation (R118G) yields an abortive enzyme (BirA*) that creates a ‘cloud’ of reactive biotinyl-AMP. This allows biotinylation of proteins on accessible lysine residues in the vicinity of the BirA*-tagged bait (estimated to be within a ~5 nM radius in one study5). Since the introduction of BioID, the proximity-dependent biotinylation enzyme toolbox has grown to include peroxidase-based enzymes (e.g., APEX2) that can label tyrosines6, biotin ligases from other species7,8 and more catalytically efficient versions of the BirA enzymes generated through directed evolution (such as TurboID and miniTurbo)9. A key feature of proximity-dependent biotinylation approaches is that spatial information is captured in living cells prior to lysis, through covalent labeling of the proximal proteins. In this case, organelles and protein interactions do not need to be maintained during lysis and purification, and as such a view of the ‘neighborhood’ of a protein of interest is obtained inside living cells.
By definition, proximity-dependent biotinylation provides an assessment of the distance relationship between a bait and a prey (three-dimensional space). Importantly, labelling can be carried out in a specific window of time, and the use of enzymes with labelling kinetics on the order of minutes (e.g., APEX2, miniTurbo and TurboID) allows for the design of experiments with a temporal perspective. With careful experimental design, spatio-temporal relationships may therefore be decoded through a variety of data analysis approaches.
We and others have begun employing proximity-dependent biotinylation approaches to map organelles and other structures in space and time. When analyzed with tools for cell localization ontologies (such as GO cellular component), proximal preys help reveal the localization of the bait to specific compartments. However, the quantitative recovery of individual preys by two baits with superficial localization to the same structure is not necessarily identical, but rather reflects the respective organization of the baits and preys within the structure10. Preys that directly bind to a bait, or are in close proximity to it within a protein complex, tend to be more strongly labeled than more distant preys within the same structure. While there is rarely sufficient information in a single bait BioID experiments to untangle these complex relationships (also see4,11 for discussions), analyzing large datasets in aggregate has enabled reconstruction of the organization of several structures and organelles – including the centrosome-cilium12, stress granules and P-bodies10, and more recently the mitochondria13. By expanding the analysis to include baits localizing across multiple distinct compartments throughout the cell we have begun to create a global ‘proximity-map’ of a cell, localizing over 4000 proteins to distinct cellular locations14 (see humancellmap.org). Expansion of these studies to explore dynamic changes in subcellular organization will constitute a stimulating challenge for experimental design, execution and data analysis.
In recent work, our group has also explored the use of BioID baits as ‘organelle sensors’ to evaluate changes in organelle proteomes following pharmacological or genetic perturbations. BioID labelling profiles are highly reproducible across experiments, and therefore performing BioID experiments with a given ‘sensor’ under different conditions (e.g., knocking-out a gene of interest, drug treatment, differential cell growth conditions) can illuminate dynamic changes that occur on specific organelle surfaces. Leveraging this approach, we used the lysosomal R-SNARE proteins VAMP7 and VAMP8 as ‘sensors’ to identify proteins localizing to the cytosolic face of late endocytic membranes (i.e., late endosomes, lysosomes, and the product of their fusion, endolysosomes) (see Figure 1). This strategy revealed novel relationships between lysosome membrane trafficking complexes and proteins involved in nutrient signalling through the large kinase complex mTORC1 (mechanistic Target Of Rapamycin, complex 1). Our follow-up studies demonstrated an unanticipated interplay between two key mTORC1 activation pathways – namely, activation by exogenous amino acids and by lysosome-derived amino acids. Importantly, the latter pathway is implicated in the growth of Ras-driven cancer cells, which can use lysosome-derived amino acids (acquired through macropinocytosis of exogenous protein) to fuel their growth. It is likely that the concept of BioID ‘organelle sensors’ will find wider application in cell biology over the coming years.
In summary, proximity-dependent biotinylation approaches offer complementary views to fractionation approaches in spatio-temporal proteomic studies. Their expanded use will allow increasingly complex cell biological questions to be answered directly in living cells.
Legend
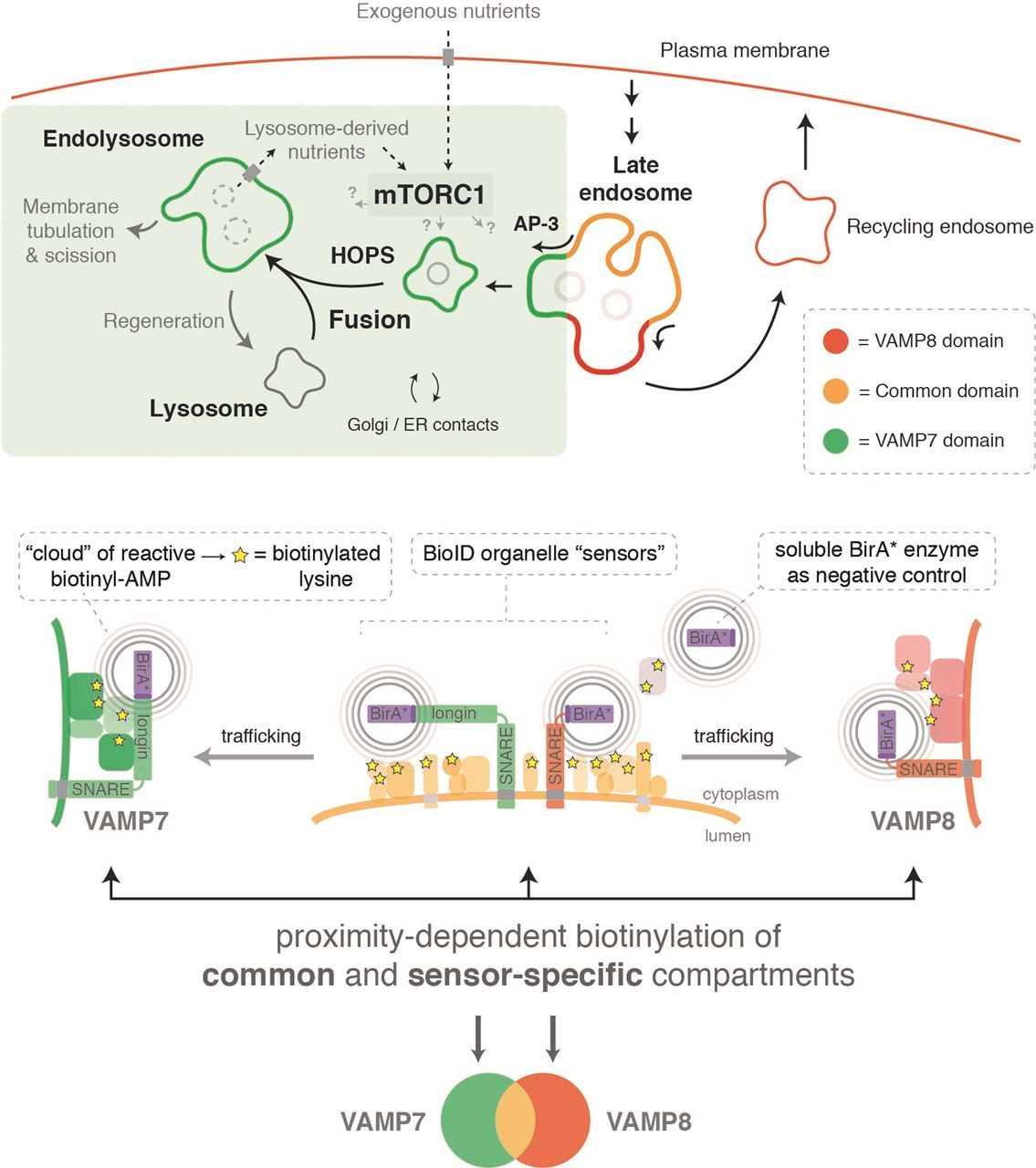
Figure 1 – The use of BioID ‘organelle sensors’ to map the surface proteomes of late endocytic organelles.
Bio

Dr. Geoffrey Hesketh is a postdoctoral fellow in Dr. Anne-Claude Gingras’ lab at the Lunenfeld-Tanenbaum Research Institute in Toronto, where he uses proteomic methods to explore how lysosomes control cell growth. He was previously a postdoctoral fellow in Dr. Paul Luzio’s lab at the Cambridge Institute for Medical Research at the University of Cambridge in the UK, where he developed his interest in lysosome biology by studying mechanisms of late endosome-lysosome fusion and recycling. Prior to this, he obtained his PhD in Dr. Jennifer Van Eyk’s lab at The Johns Hopkins University School of Medicine.
References
1. Lundberg, E. & Borner, G. H. H. Spatial proteomics: a powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 1 (2019). doi:10.1038/s41580-018-0094-y
2. Prinz, W. A., Toulmay, A. & Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 21, 7–24 (2020).
3. Roux, K. J., Kim, D. I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–10 (2012).
4. Samavarchi-Tehrani, P., Samson, R. & Gingras, A.-C. Proximity Dependent Biotinylation: Key Enzymes and Adaptation to Proteomics Approaches. Mol. Cell. Proteomics 19, 757–773 (2020).
5. Kim, D. I. et al. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. U. S. A. 111, E2453-61 (2014).
6. Lam, S. S. et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015).
7. Kim, D. I. et al. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 27, 1188–96 (2016).
8. Ramanathan, M. et al. RNA-protein interaction detection in living cells. Nat. Methods 15, 207–212 (2018).
9. Branon, T. C. et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018).
10. Youn, J.-Y. et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 517–532 (2018). doi:10.1016/j.molcel.2017.12.020
11. Gingras, A.-C., Abe, K. T. & Raught, B. Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 48, 44–54 (2019).
12. Gupta, G. D. et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 163, 1483–1499 (2015).
13. Antonicka, H. et al. A High-Density Human Mitochondrial Proximity Interaction Network. Cell Metab. 32, 479-497.e9 (2020).
14. Go, C. et al. A proximity biotinylation map of a human cell. (2019). doi:10.1101/796391


.png)












