Authors:
Yun-En Chung1 and Mathieu Lavallée-Adam1
1Department of Biochemistry, Microbiology and Immunology and Ottawa Institute of Systems Biology, University of Ottawa, Ottawa, Ontario, Canada
Mass spectrometry-based proteomics data analysis has never been more exciting. The combination of computational hardware improvements and a wide diversity of instruments and experimental techniques has created a gigantic playground for computational researchers and software developers. In recent years, one attraction at this playground has gained a lot of attention from both academia and industry: real-time analysis of proteomics data.
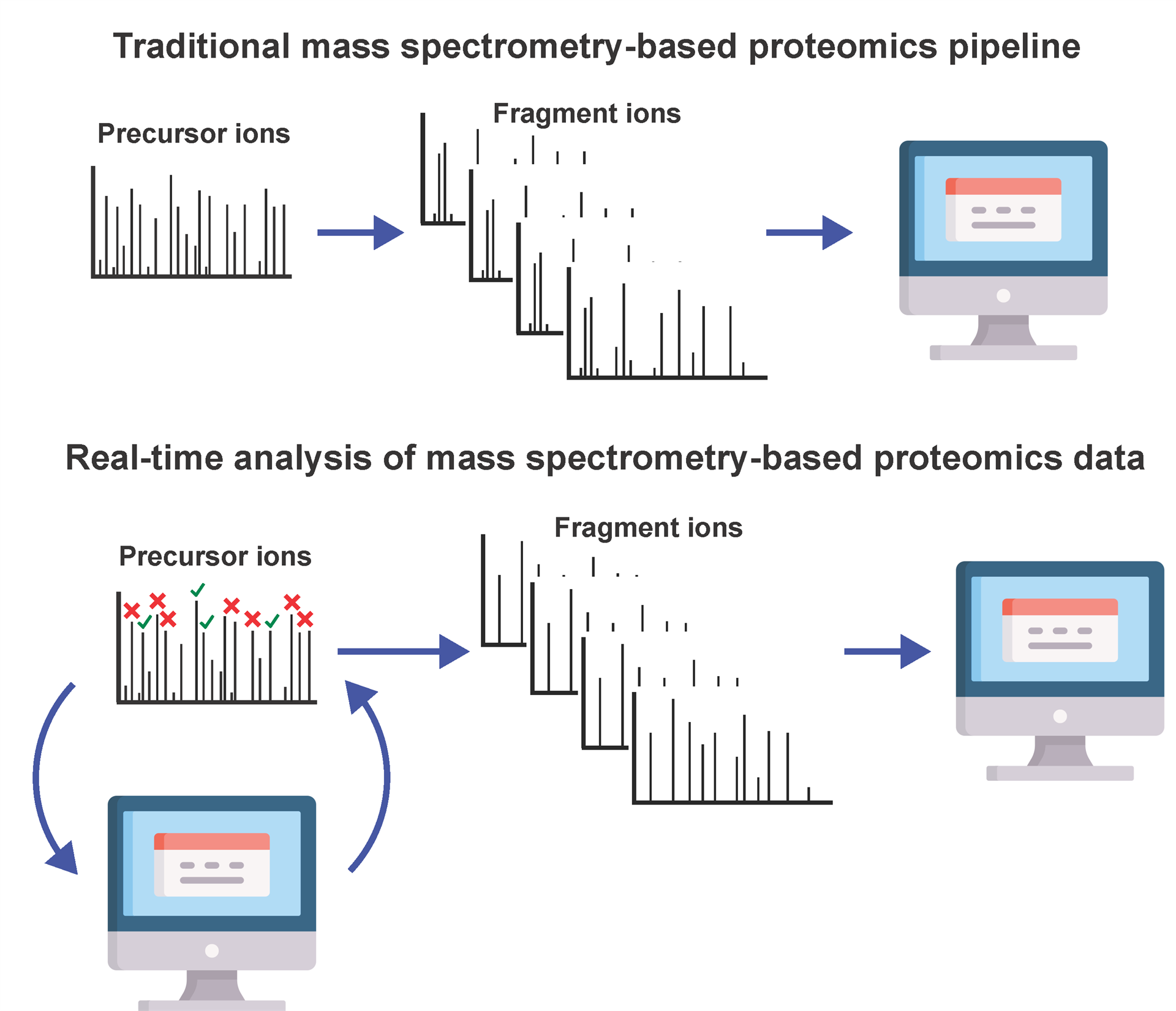
In a traditional mass spectrometry-based proteomics experiment, tens of thousands of mass spectra are collected for a biological sample. After the conclusion of the experiment these mass spectra are then inputted into software packages for peptide and protein identification and quantification. Hence, since biological information is inferred solely through post-hoc analysis, the mass spectrometry experiment is mostly running blind and does not adapt in real-time based on the biological data that is being acquired.
Improvements in computational hardware and the recent availability of Application Programming Interfaces (APIs) enabling mass spectrometry data analysis during proteomics experiments have paved the way to the design of a new family of algorithms and software packages performing the real-time analysis of mass spectrometry data. Tools such as the Thermo Fisher Scientific Instrument API (IAPI)1 and the Bruker Parallel database Search Engine in Real-time (PaSER)2 are enabling the design of analyses that accelerate data processing, help diagnose problems with instrumentation and enhance the characterization capabilities of mass spectrometry.
One of the early modern applications of real-time mass spectrometry data analysis is the on-the-fly quality assessment of mass spectrometry experiments. Instrument performance drop or malfunction are often only identified after post-hoc data analysis. Such a late discovery results in a waste of time and resources that are used to acquire unusable or subpar data. The QC-ART approach3 has been developed to evaluate instrument performance in near real-time and allow for immediate intervention. QC-ART ensures consistent high-quality data collection and the rapid detection of instrumentation problems.
Since the early beginnings of mass spectrometry-based proteomics, data acquisition remained an extremely active research topic. Still today, new acquisition techniques are being developed to supplement the current families of approaches including data-dependent acquisition4, data-independent acquisition5 and targeted methods6,7. Traditionally, an instrument would apply the same acquisition strategy (precursor ion selection algorithm, scan window size, …) for the entirety of an experiment. This standard acquisition method works reasonably well in common proteomics use cases. However, since the instrument does not consider the biological relevance of the data it is acquiring, a significant proportion of this data does not translate into meaningful biological discoveries.
An excellent example of this is how real-time database search for peptide identification can support the selection of peptides for quantification with isobaric labeling. It was previously shown that MS3 spectra lead to more accurate quantification using tandem mass tag reporter ions than MS2 spectra8. However, acquiring MS3 spectra is resource intensive. It is therefore important to acquire MS3 spectra for data that is biologically relevant. Orbiter was therefore developed to identify peptides in real-time from MS2 spectra with a database search method9. Orbiter then only acquires MS3 spectra from MS2 spectra that yield a confident peptide identification and therefore optimizes resource usage for protein quantification.
Other groups developed software packages to identify peptides in real-time10,11, while McQueen et al. presented a pseudo real-time approach that paused the experiment to adjust future data acquisition based on such peptide identifications. Inspired by these methods, our team proposed that the real-time identification of peptides and proteins can be used to guide mass spectrometry data acquisition in order to optimize resource usage and maximize protein identifications. Indeed, our computational approach, named MealTime-MS12, uses real-time database search to identify peptides and supervised learning to assess the confidence of protein identifications. MealTime-MS then uses confident protein identifications to generate an exclusion list preventing the acquisition of tandem mass spectra for peptide ions that are expected to belong to proteins that were already identified in the mass spectrometry run. MealTime-MS showed that up to 33% of the mass spectra collected in traditional experiments could be safely ignored with minimal losses of proteins identified compared to standard experiments and that these mass spectra could be repurposed for the identification of additional proteins.
Alternatively, real-time analysis of mass spectrometry data has demonstrated its utility in targeted proteomics. In a typical targeted proteomics experiment, specific elution time windows need to be determined for targeted peptides. Due to run-to-run technical variation, the size of these scheduled windows must be kept relatively large to ensure the instrument encounters these peptides, thereby limiting the number of possible targets. MaxQuant.Live presented a solution via real-time recognition of precursor ions13. The algorithm uses the retention time, mass-to-charge ratio, and intensity of the precursor ions encountered to predict and therefore select those that should be targeted for quantification. This approach enabled the targeting of over 25,000 peptides in a single mass spectrometry run.
Real-time analysis of mass spectrometry-based proteomics data has also demonstrated its clinical applications. Devices such as the MasSpec Pen demonstrated how a small handheld device can be used to rapidly detect features including lipids, metabolites and proteins in human tissue. Such features can be used as biomarkers to diagnose in real-time whether tissues are cancerous or healthy.
After reading about these applications, we would like you to join the conversation on Twitter by answering our poll question here and letting us know where the future of real-time proteomics data analysis sits:
Twitter Poll:
In which area do you think real-time analysis of mass spectrometry-based proteomics data will have the greatest impact in the future:
- Protein ID
- Protein Quantification
- Clinical Applications
- Other (write in replies)
Figure 1. Graphical representation of the traditional mass spectrometry-based proteomics pipeline, where acquired data is analyzed after the completion of the experiment and of a pipeline integrating real-time data analysis to adjust mass spectrometry data acquisition during the experiment.
Computer Icon created by Freepik - Flaticon: https://www.flaticon.com/free-icons/course.
Bios:
Yun-En Chung:
 Yun-En Chung is an undergraduate student in Translational and Molecular Medicine and researcher in Dr. Mathieu Lavallée-Adam’s lab at the University of Ottawa. His research focuses on the development of software packages to guide mass spectrometry experiments in real-time to improve data acquisition efficiency. His publication on the real-time identification of proteins in mass spectrometry data was recognized as the best paper from a Master’s or Undergraduate student at the Ottawa Institute of Systems Biology in 2020. He also received several awards for his presentations, including a 2nd place for his oral presentation at the Undergraduate Research Opportunities Program Seminar day at the University of Ottawa and an honorable mention for his poster at the American Society for Mass Spectrometry annual conference in 2022. Yun-En’s research is funded by awards from the Natural Sciences and Engineering Research Council of Canada and Mitacs.
Yun-En Chung is an undergraduate student in Translational and Molecular Medicine and researcher in Dr. Mathieu Lavallée-Adam’s lab at the University of Ottawa. His research focuses on the development of software packages to guide mass spectrometry experiments in real-time to improve data acquisition efficiency. His publication on the real-time identification of proteins in mass spectrometry data was recognized as the best paper from a Master’s or Undergraduate student at the Ottawa Institute of Systems Biology in 2020. He also received several awards for his presentations, including a 2nd place for his oral presentation at the Undergraduate Research Opportunities Program Seminar day at the University of Ottawa and an honorable mention for his poster at the American Society for Mass Spectrometry annual conference in 2022. Yun-En’s research is funded by awards from the Natural Sciences and Engineering Research Council of Canada and Mitacs.
Mathieu Lavallée-Adam:
 Mathieu Lavallée-Adam is an Associate Professor at the University of Ottawa in the Department of Biochemistry, Microbiology and Immunology and is affiliated to the Ottawa Institute of Systems Biology. He obtained a B.Sc. in Computer Science and a Ph.D. in Computer Science, Bioinformatics option, from McGill University and performed his postdoctoral research at The Scripps Research Institute. His research focuses on the development of statistical and machine learning algorithms for the analysis of mass spectrometry-based proteomics data and protein-protein interaction networks. Dr. Lavallée-Adam is a recipient of the John Charles Polanyi Prize in Chemistry, rewarding the impact of his bioinformatics algorithms on the mass spectrometry community and was named Early Career Researcher of the Year by the Ottawa Institute for Systems Biology in 2021. He is also Co-Chair of the HUPO Early Career Researcher Initiative and a member of the HUPO Executive Committee, in which he develops training activities and advocates for junior investigators in proteomics and organize events highlighting their research on the international stage.
Mathieu Lavallée-Adam is an Associate Professor at the University of Ottawa in the Department of Biochemistry, Microbiology and Immunology and is affiliated to the Ottawa Institute of Systems Biology. He obtained a B.Sc. in Computer Science and a Ph.D. in Computer Science, Bioinformatics option, from McGill University and performed his postdoctoral research at The Scripps Research Institute. His research focuses on the development of statistical and machine learning algorithms for the analysis of mass spectrometry-based proteomics data and protein-protein interaction networks. Dr. Lavallée-Adam is a recipient of the John Charles Polanyi Prize in Chemistry, rewarding the impact of his bioinformatics algorithms on the mass spectrometry community and was named Early Career Researcher of the Year by the Ottawa Institute for Systems Biology in 2021. He is also Co-Chair of the HUPO Early Career Researcher Initiative and a member of the HUPO Executive Committee, in which he develops training activities and advocates for junior investigators in proteomics and organize events highlighting their research on the international stage.
References:
1. Scientific, T. F. Thermo Fisher Scientific IAPI GitHub. (2022). Available at: https://github.com/thermofisherlsms/iapi.
2. Bruker. PaSER 2022. (2022).
3. Stanfill, B. A., Nakayasu, E. S., Bramer, L. M., Thompson, A. M., Ansong, C. K., Clauss, T. R., Gritsenko, M. A., Monroe, M. E., Moore, R. J., Orton, D. J., Piehowski, P. D., Schepmoes, A. A., Smith, R. D., Webb-Robertson, B.-J. M., Metz, T. O. & TEDDY Study Group. Quality Control Analysis in Real-time (QC-ART): A Tool for Real-time Quality Control Assessment of Mass Spectrometry-based Proteomics Data. Mol. Cell. Proteomics 17, 1824–1836 (2018).
4. Liu, H., Sadygov, R. G. & Yates, J. R. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–201 (2004).
5. Venable, J. D., Dong, M.-Q., Wohlschlegel, J., Dillin, A. & Yates, J. R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 1, 39–45 (2004).
6. Kuhn, E., Wu, J., Karl, J., Liao, H., Zolg, W. & Guild, B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics 4, 1175–86 (2004).
7. Anderson, L. & Hunter, C. L. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–88 (2006).
8. Ting, L., Rad, R., Gygi, S. P. & Haas, W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–40 (2011).
9. Schweppe, D. K., Eng, J. K., Yu, Q., Bailey, D., Rad, R., Navarrete-Perea, J., Huttlin, E. L., Erickson, B. K., Paulo, J. A. & Gygi, S. P. Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J. Proteome Res. 19, 2026–2034 (2020).
10. Bailey, D. J., Rose, C. M., McAlister, G. C., Brumbaugh, J., Yu, P., Wenger, C. D., Westphall, M. S., Thomson, J. A. & Coon, J. J. Instant spectral assignment for advanced decision tree-driven mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 109, 8411–6 (2012).
11. Graumann, J., Scheltema, R. A., Zhang, Y., Cox, J. & Mann, M. A framework for intelligent data acquisition and real-time database searching for shotgun proteomics. Mol. Cell. Proteomics 11, M111.013185 (2012).
12. Pelletier, A. R., Chung, Y.-E., Ning, Z., Wong, N., Figeys, D. & Lavallée-Adam, M. MealTime-MS: A Machine Learning-Guided Real-Time Mass Spectrometry Analysis for Protein Identification and Efficient Dynamic Exclusion. J. Am. Soc. Mass Spectrom. 31, 1459–1472 (2020).
13. Wichmann, C., Meier, F., Winter, S. V., Brunner, A.-D., Cox, J. & Mann, M. MaxQuant.Live enables global targeting of more than 25,000 peptides. bioRxiv 443838 (2018). doi:10.1101/443838


.png)












