Niphakis, M.J. et al. Cell 161, 1668–1680 (18 June 2015)
News in Science (HUPOST Vol. 5, Q3 September 2015)
Lipids are a class of hydrocarbon-containing natural molecules; important examples include some vitamins (A, D, E), hormones (estrogen, testosterone), neurotransmitters (endocannabinoids) and components of fat (triglycerides, cholesterol). Their biological functions to regulate cell physiology and disease are often mediated through interactions with proteins. Despite the fact that a substantial number of drug targets are lipid-binding proteins, it has been challenging to establish a global portrait of how lipids interact with proteins in cells.
Benjamin F. Cravatt’s team at The Scripps Research Institute (TSRI) has devised a powerful chemical proteomic method to map the human lipid-binding proteome. In this study, Niphakis and colleagues described a set of fatty acid–based chemical probes combined with quantitative mass spectrometry to provide the first global portrait of lipid-protein interactions in human cells. Their results revealed a surprisingly large number of lipid-protein interactions from diverse functional classes, including some interactions which are already targeted by existing drugs and, most importantly, many novel lipid-binding proteins. The probes can also be used to identify the targets of drugs that disrupt lipid-protein interactions in cells, pointing to the broad potential of lipid-binding proteins for ligand/drug development.
“Traditionally, scientists have theorized fairly narrow limits for the types of proteins that can be targeted with small-molecule ligands, but our results suggest that those boundaries are much wider,” said Cravatt (adapted from News Release of The Scripps Research Institute)
This study was reported in the journal of Cell on June 18, 2015.
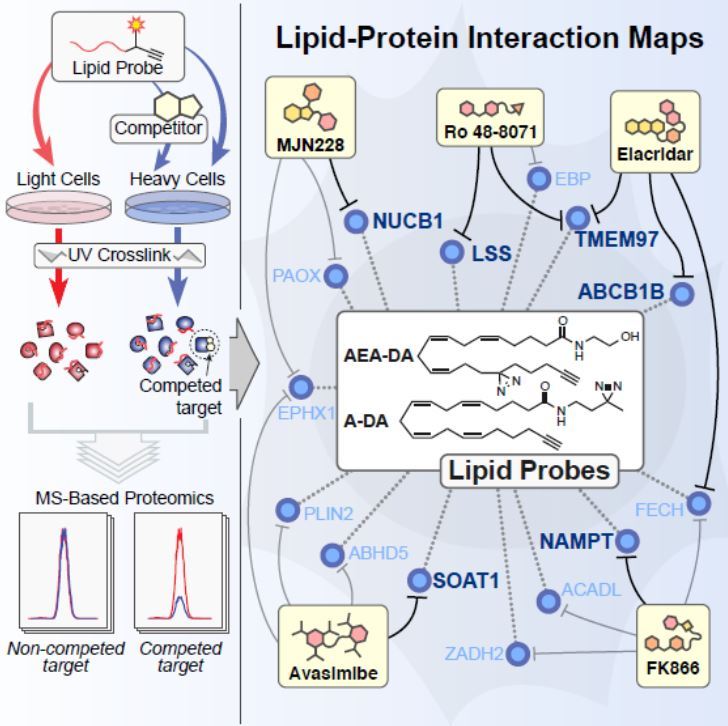 Figure provided by Benjamin F. Cravatt
Figure provided by Benjamin F. Cravatt
http://www.cell.com/cell/abstract/S0092-8674(15)00635-2


.png)












